
sc::MBPT2 Class Reference
The MBPT2 class implements several second-order perturbation theory methods. More...
#include <mbpt.h>
Inheritance diagram for sc::MBPT2:
Public Methods | |
| MBPT2 (StateIn &) | |
| MBPT2 (const Ref< KeyVal > &) | |
| The KeyVal constructor. More... | |
| ~MBPT2 () | |
| void | save_data_state (StateOut &) |
| Save the base classes (with save_data_state) and the members in the same order that the StateIn CTOR initializes them. More... | |
| Ref< SCF > | ref () |
| double | ref_energy () |
| double | corr_energy () |
| RefSCVector | ref_energy_gradient () |
| RefSCVector | corr_energy_gradient () |
| int | nelectron () |
| Returns the number of electrons. | |
| RefSymmSCMatrix | density () |
| Returns the SO density. | |
| int | spin_polarized () |
| Return 1 if the alpha density is not equal to the beta density. | |
| int | gradient_implemented () const |
| int | value_implemented () const |
| Information about the availability of values, gradients, and hessians. | |
| void | symmetry_changed () |
| Call this if you have changed the molecular symmetry of the molecule contained by this MolecularEnergy. | |
| void | obsolete () |
| Marks all results as being out of date. More... | |
| void | print (std::ostream &o=ExEnv::out0()) const |
| Print information about the object. | |
Protected Methods | |
| void | init_variables () |
| void | compute () |
| Recompute at least the results that have compute true and are not already computed. More... | |
| void | eigen (RefDiagSCMatrix &vals, RefSCMatrix &vecs, RefDiagSCMatrix &occs) |
| void | compute_hsos_v1 () |
| distsize_t | compute_v2_memory (int ni, int nfuncmax, int nbfme, int nshell, int ndocc, int nsocc, int nvir, int nproc) |
| void | compute_hsos_v2 () |
| void | compute_hsos_v2_lb () |
| int | compute_cs_batchsize (size_t mem_static, int nocc_act) |
| distsize_t | compute_cs_dynamic_memory (int ni, int nocc_act) |
| int | make_cs_gmat (RefSymmSCMatrix &Gmat, double *DPmat) |
| int | make_cs_gmat_new (RefSymmSCMatrix &Gmat, const RefSymmSCMatrix &DPmat) |
| void | form_max_dens (double *DPmat, signed char *maxp) |
| int | init_cs_gmat () |
| void | done_cs_gmat () |
| int | make_g_d_nor (RefSymmSCMatrix &Gmat, double *DPmat, const double *mgdbuff) |
| void | cs_cphf (double **scf_vector, double *Laj, double *eigval, RefSCMatrix &P2aj) |
| void | s2pdm_contrib (const double *intderbuf, double *PHF, double *P2AO, double **hf_ginter, double **ginter) |
| void | hcore_cs_grad (double *PHF, double *PMP2, double **hf_ginter, double **ginter) |
| void | overlap_cs_grad (double *WHF, double *WMP2, double **hf_ginter, double **ginter) |
| void | compute_cs_grad () |
Protected Attributes | |
| Ref< SCF > | reference_ |
| Ref< MemoryGrp > | mem |
| int | nfzc |
| int | nfzv |
| size_t | mem_alloc |
| double | cphf_epsilon_ |
| int | eliminate_in_gmat_ |
| const double * | intbuf_ |
| Ref< TwoBodyInt > | tbint_ |
| Ref< TwoBodyInt > * | tbints_ |
| Ref< TwoBodyDerivInt > * | tbintder_ |
| int | nbasis |
| int | noso |
| Ref< MessageGrp > | msg_ |
| int | nvir |
| int | nocc |
| int | nsocc |
| Ref< ThreadGrp > | thr_ |
| int | dynamic_ |
| int | max_norb_ |
| int * | symorb_irrep_ |
| int * | symorb_num_ |
| char * | method_ |
| char * | algorithm_ |
| int | do_d1_ |
| int | do_d2_ |
| int | nfuncmax |
| double | hf_energy_ |
| RefSCVector | hf_gradient_ |
| double | restart_ecorr_ |
| int | restart_orbital_v1_ |
| int | restart_orbital_memgrp_ |
Detailed Description
The MBPT2 class implements several second-order perturbation theory methods.
Constructor & Destructor Documentation
|
|
The KeyVal constructor.
|
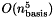 part of the calculation. Only energies can be computed. This is recommended only for computations involving large molecules (where the transformation is dominant) on very many processors (approaching the number of shells).
part of the calculation. Only energies can be computed. This is recommended only for computations involving large molecules (where the transformation is dominant) on very many processors (approaching the number of shells).
Member Function Documentation
|
|
Recompute at least the results that have compute true and are not already computed. This should only be called by Result's members. Implements sc::Compute. |
|
|
Marks all results as being out of date. Any subsequent access to results will cause Compute::compute() to be called. Reimplemented from sc::Wavefunction. |
|
|
Save the base classes (with save_data_state) and the members in the same order that the StateIn CTOR initializes them. This must be implemented by the derived class if the class has data. Reimplemented from sc::Wavefunction. |
The documentation for this class was generated from the following file:
Generated at Fri Jan 10 08:15:28 2003 for MPQC 2.1.3 using the documentation package Doxygen 1.2.14.